
O Lisossoma
Lisossoma | Lisossoma – O crescimento celular e o metabolismo envolvem processos fisiológicos associados à manutenção da homeostasia nos tecidos. O metabolismo envolve a degradação de componentes celulares (como proteínas, ácidos nucleicos e hidratos de carbono) e a reconstituição destes produtos degradados em novas macromoléculas. Os lisossomas desempenham um papel fundamental nestes processos e fornecem um ambiente interno adequado (pH baixo) para manter muitas enzimas hidrólases ácidas. Estas enzimas hidrólases actuam na degradação de macromoléculas em elementos que são posteriormente reutilizados pelas células ou eliminados do organismo. Quando uma destas enzimas se encontra em falta ou existe em pequenas quantidades, verifica-se um aumento progressivo de moléculas não degradadas no interior dos lisossomas.
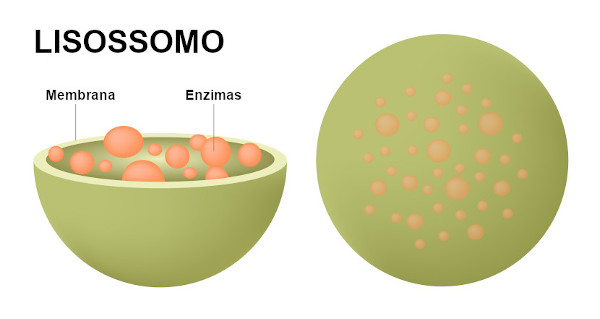
São organelas citoplasmáticas que fazem parte do sistema de endomenbrana, envolvidos por uma membrana lipoproteica, que diferem no tamanho e forma. No seu interior contém enzimas hidrolíticas que intervém no mecanismo de digestão célula.
Os lisossomos são formados no Complexo de Golgi (outra importante organela presente no citoplasma). As enzimas são produzidas no RER e migram para os dictiossomos, sendo identificadas e enviadas para uma região especial do aparelho de Golgi, onde são empacotadas e libertadas sob a forma de pequenas bolsas.
– Fazer a degradação e digestão de partículas originárias do meio exterior às células;
– Reciclar (função de renovação celular) outras organelas celulares que estão envelhecidas. Este processo é conhecido como autofagia.
São organelas citoplasmáticas que fazem parte do sistema de endomenbrana, envolvidos por uma membrana lipoproteica, que diferem no tamanho e forma. No seu interior contém enzimas hidrolíticas que intervém no mecanismo de digestão célula.
As doenças lisosomais, um grupo de mais de 50 doenças:
As Lipidoses: Austin, Fabry, Faber, Gaucher (tipo I, II e III), Gangliosidose GM1 (Landing), Krabbe, Leucodistrofia Metacromática, Sandhoff, Niemann – Pick (A/B e C), Schindler-Kanzaki, Tay-Sachs, Wolman, Doença de sobrecarga em ésteres de colesterol.
A Glicogenose tipo II: Doença de Pompe.
As Oligossacaridoses e Glicoproteinoses: Aspartilglucosaminuria, Fucosidose, Alfa- e Beta-Manosidoses, Mucolipidose tipo II (I-Cell), Mucolipidose tipo III (Pseudo Hurler), Mucolipidose tipo IV, Sialidose, Galactosidose.
Anomalias da transferência lisosomal: Cistinose, Doença de Danon, Doença de Salla (sobrecarga em ácido siálico livre).
As Mucopolissacaridoses (MPS): MPS I (Hurler, Hurler-Scheie, Scheie), MPS II (Hunter), MPS III (Sanfilippo A, B, C e D), MPS IV (Morquio A e B), MPS VI (Maroteaux-Lamy), MPS VII (Sly), MPS IX.
Lipofuscinoses ceróides neuronais: CLN tipo 1 a 10.
Outras doenças lisosomais: Pycnodysostosis, Síndrome de Papillon-Lefèvre, Síndrome de Chediak-Higashi.